

QGP Aggregation Pipeline
Version 1.0
The samples that PASS the pre-aggregation QC pipeline (per sample QC) are aggregated into one multi-sample VCF file. Samples are aggregated using Dragen Iterative gVCF genotyper. To accelerate the aggregation and the downstream analyses (parallel processing), the genome is divided into 100 shards.
The pipeline produces one multi-sample VCF file per shard, as well as a global multi-sample VCF file that concatenates all shards.
Alongside the multi-sample VCF generation, we also perform the following analyses:
Functional annotation
Each msVCF (global or per shard) is annotated using Ensembl Variant Effect Predictor (VEP) tool, version 112. In addition to VEP default annotations, we added the below databases:
- ClinVar v20240805
- HGMD Pro v2024.2
- GnomAD genomes v4.0
- GnomAD exomes v4.0
High quality dataset of SNPs (HQ) - (cohort-level QC)
We initially converted the multi-sample VCF (msVCF) into Bed/Bim/Fam format using the Plink 1.9 tool. These files were then utilized to generate a high-quality set of SNPs, also using Plink 1.9. The SNPs were selected based on the following criteria:
- Include autosomal, bi-allelic SNPs only
- Common variants only (MAF > 5%)
- Remove variants with missingness rate > 10%
- Remove samples with missingness rate > 5%
- LD prune with an r2 of 0.1, 500kb window
- Hardy Weinberg Equilibrium pHWE < 1e-5
Principal Components and genetically inferred relatedness
Using the HQ SNPs, we run PCA using the GCTA tool (v1.94). The first 20 PCs are provided. We also generated a pairwise kinship matrix using KING tool (v2.2.7), in addition to a list of unrelated samples (up to 2nd-degree).
Hail matrix
The global msVCF, is also converted into Hail (v0.2) MatrixTable format. This matrix can be imported to perform highly parallelized analysis.
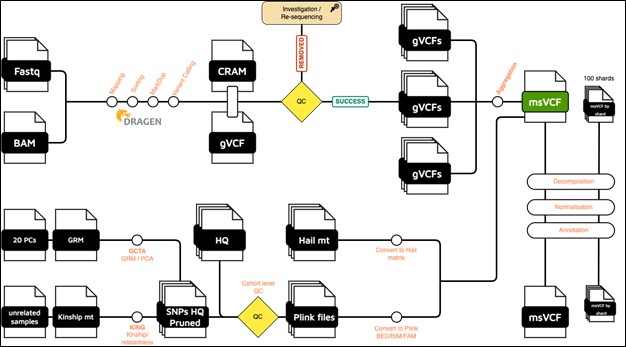
Figure 1: Global overview of the QPHI analysis pipeline
QGP Pre-Aggregation QC pipeline (individual level)
Version 1
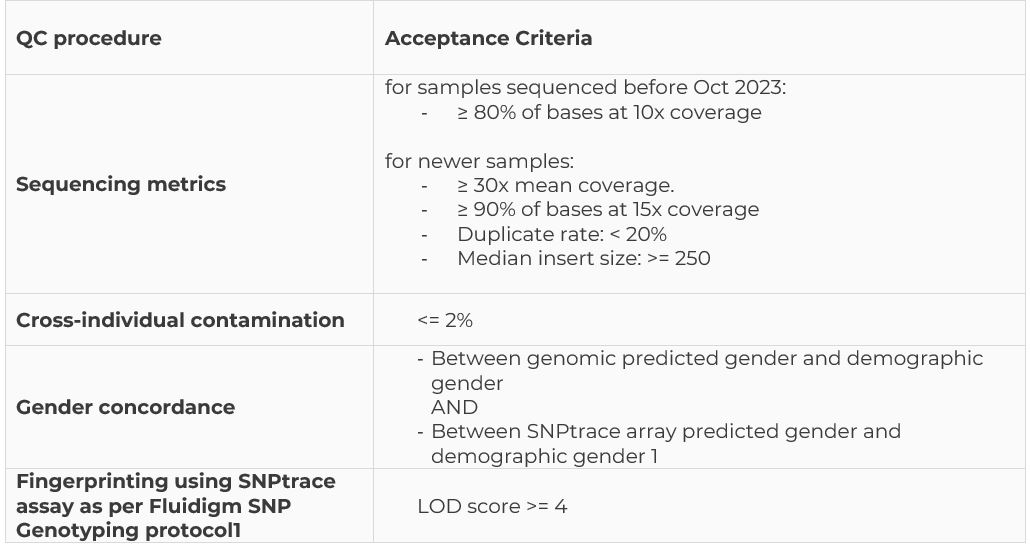
Each WGS sample is tested independently through this pipeline. If a sample fails the test, it is excluded from the aggregation process and will not be included in the final release. These tests are designed to identify sample swaps, cross-individual contamination, and errors in sample preparation or sequencing. A detailed list of specific QC processes is provided in the table below:
QGP Germline Whole Genome Primary & Secondary analyses pipeline
Version 1
Genomic raw data is processed using the Dragen solution.

All callers from the Dragen DNA pipeline are activated.
This pipeline takes two types of input files:
- BAM files: for the previously analyzed samples (with Sentieon tool). The header of these BAM files has been modified to update the sample name field (according to the new QPHI Sample IDs) and other metadata related to the sequencing run.
- Fastq files: for newly sequenced samples.
QPHI-Qatari (QPHI-P-Q)
Version 1 (25k release)
Changelog
- Lossless CRAM replaces the BAM format for reads alignement files
- Sample IDs in CRAM and VCF files are updated to keep only the QPHI unique ID
- New Individual and cohort-level Quality Control (QC) steps introduced in the QGP Pre-Aggregation QC pipeline V1, to remove samples with sequencing issues
- New file formats are delivered for each release (Plink, Hail, PCs, Relatedness, etc.)
- New variants callers (SV, CNV, PGx, SMN, HLA, STRs, etc.)
- New iterative approache for cohort gVCF aggregation, introduced in the QGP Aggregation pipeline v1.0
- New releases file structure for individual and cohort-level files
- New annotation databases (HGMD pro) added to the msVCF, in addition to standard public annotation databases
Genomes
Genomic data release Version 1 provides germline genomes (WGS) of 24,838 healthy participants, from Qatar BioBank (QBB) population-based cohort. This release is generated using the Aggregation pipeline (v1.0).
As part of this release, we’re providing several data formats, in addition to the multi-sample VCF file, to make it easier and faster for researchers to analyze the cohort. Below are the files included:
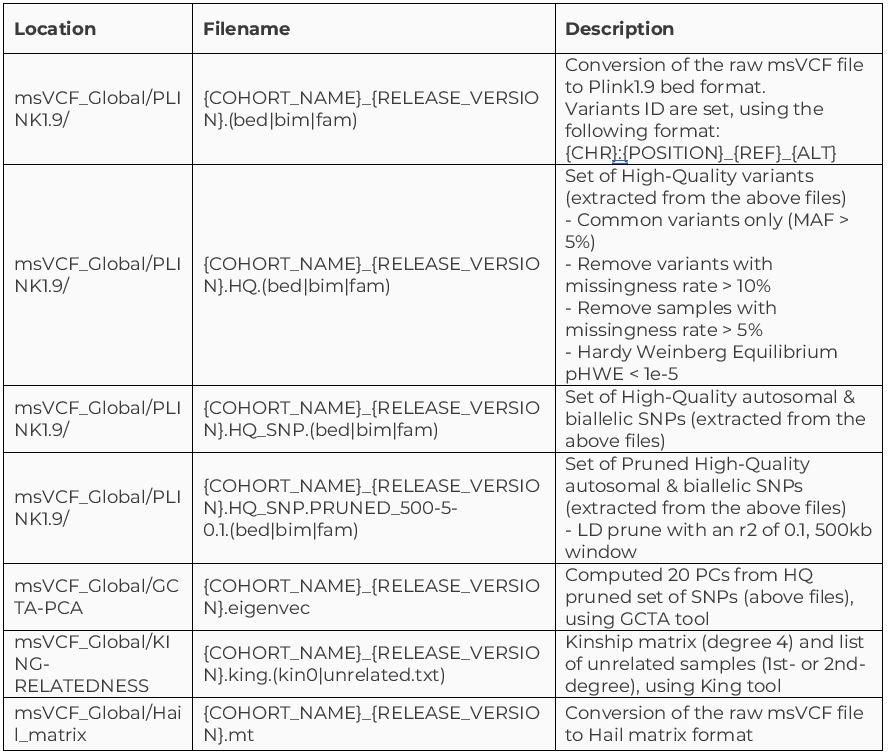
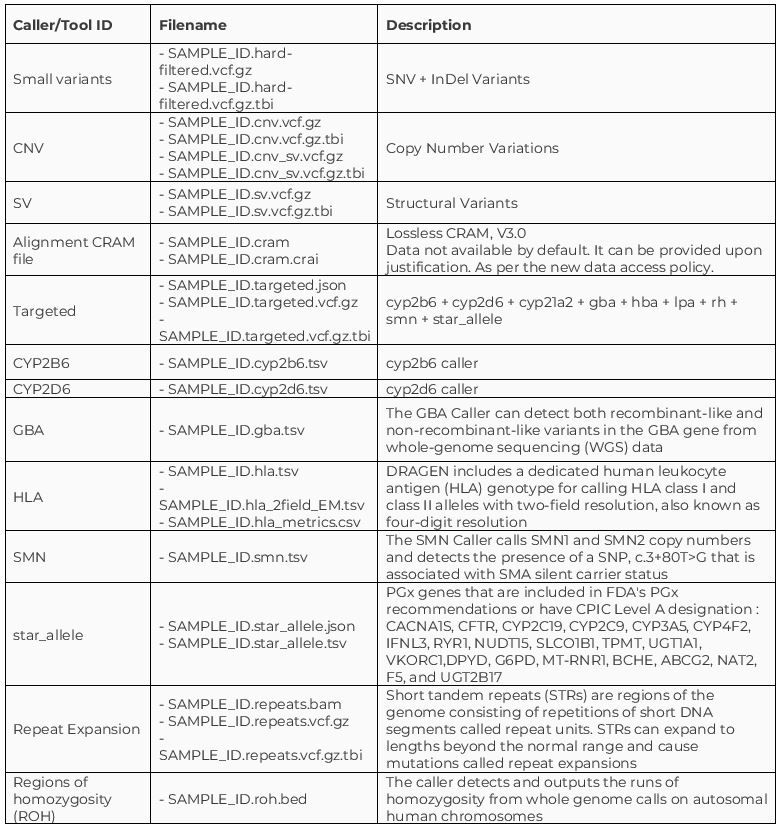
In addition to the cohort-level files produced by the Aggregation pipeline, each participant has the following genomic file types, generated by different callers:
Proteomics :
Number of participants included in the 25k genomic release and having proteomics data : 2817
Metabolomics
Number of participants included in the 25k genomic release and having metabolomics data : 2877